
代谢组学是一个主要的组学课题,在代谢特征和生物标志物的临床应用和基础研究中都占有重要地位。不幸的是,相关研究受到许多外部因素造成的批次效应的挑战。在过去十年中,深度学习技术已成为数据科学中的主要工具,人们可以从已知批次训练诊断网络,然后将其推广到新批次。然而,批次效应不可避免地阻碍了这种努力,因为所考虑的两个批次可能高度不匹配。
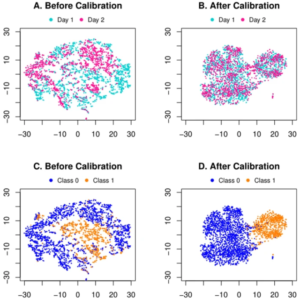
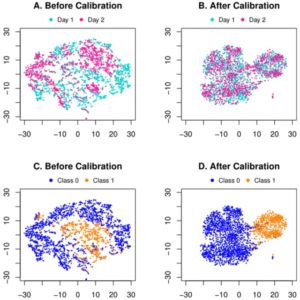
上海交通大学和上海科技大学的研究人员提出了一个端到端的深度学习框架,用于联合批次效应去除,然后对代谢组学数据进行分类。研究人员首先在公共 CyTOF 数据集上验证提出的深度学习框架作为模拟实验。他们还直观地比较了 t-SNE 分布,并证明该方法有效地消除了潜在空间中的批处理效应。
然后,对于私有 MALDI MS 数据集,实现了最高的诊断准确性,与之前最先进的方法相比,平均提高了约 5.1 ~ 7.9%。两个实验均得出结论,该方法在分类方面的表现明显优于传统方法,这得益于有效去除批次效应。
该研究以「Joint deep learning for batch effect removal and classification toward MALDI MS based metabolomics」为题,于 2022 年 7 月 10 日发布在《BMC Bioinformatics》。

代谢组学寻找复杂生物样品的定量描述,并将疾病的临床观察与代谢物的时间波动联系起来。通过测量和模拟生物样本中的代谢变化,代谢组学可以深入了解饮食、疾病和疗法的影响。许多新的代谢组学技术出现,现在可以有效地分析大量数据。例如,基质辅助激光解吸/电离质谱(MALDI MS)提供快速的处理速度和低样品消耗量。
图示:患者的公共 CyTOF 数据的可视化。(来源:论文)
同时,飞行时间流式细胞仪(CyTOF)是一种新颖的单细胞分析技术,将数据维度扩展到同时测量 40 + 细胞参数。因此,它已被应用于跟踪反映各种细胞属性的生物标志物的表达水平。
虽然非靶向代谢组学具有编码复杂生物样品的能力,但通常必须使用复杂的数据解释技术来促进临床应用。许多传统研究在人群水平上建立了统计差异。
然而,为了个体化诊断和治疗,将他们的发现推广到样本或受试者水平并不容易。鉴于机器学习,尤其是深度学习在过去几年的快速发展,有一种趋势是采用这些先进的工具从非靶向代谢组学数据中搜索生物标志物,然后建立适用于个体患者的数据驱动的疾病模型。
将机器学习模型推广到真实案例代谢组学数据的一个主要挑战是批处理效应,这几乎是不可避免的。确切地说,批次效应解释了在实验条件下表现不同但与所考虑的感兴趣的生物变量无关的测量。批次效应的起源是广泛的,包括不同的平台、同一样本的不同试剂、不同的时间点获取数据等。因此,有必要在这些代谢组学场景中抑制批次效应。
批次效应的瓶颈在过去引起了激烈的研究。有两种抑制批次效应的传统方法,即位置尺度(LS)和矩阵分解(MF)。例如,ComBat 是一种流行的 LS 方法。它采用贝叶斯框架对数据进行建模,通过独立参数化每个批次和每个特征的位置和规模。其他 LS 方法,包括距离加权鉴别(DWD)、单向方差分析(ANOVA)方法和 Ratio_G,假设每个批次的数据分布正常,并相应地调整不同批次的分布。然而,LS 方法中的假设可能被过度简化,以将复杂的批次效应视为加法和乘法分量。
作为 LS 方法的替代方法,代理变量分析 (SVA) 提供了一种消除批次效应的 MF 方法。MF 方法假设由批次效应引起的数据变化与目标标签无关。这样,数据就可以分解成两部分,分别对应 batch effect的失真和遗漏的部分。然而,MF 方法中的建模依赖于对批次效应和数据标签独立性的假设,这在实践中可能并不总是有效。
通常,在非靶向代谢组学诊断领域,需要构建一个判别模型,该模型可以训练一批源数据并将其应用于预测目标批数据的标签。Ratio_G 方法调整数据以增强标签预测。fSVA 方法还能够预测未见样本的标签。
在 fSVA 中,首先使用 SVA 来校准训练数据集中的批次效应。然后,利用在训练数据集上估计的概率权重和系数来消除新测试样本中的批次效应。在校准数据集上训练的分类器最终可以应用于预测。然而,矩阵分解有时可能会减少归因于批次效应的数据变化,但代价是减少疾病组和对照之间的差异,这反过来又会降低后续分析中的分类准确性。
近几十年来,涉及基因组学、转录组学、蛋白质组学、代谢组学和放射组学的高通量组学数据的计算分析变得很流行。考虑到许多测量(对应于特征维度)和通常少量的样本(或样本量),这显然是对机器学习的挑战。
另一方面,最近深度学习的飞跃提供了一种前所未有的工具来克服这些障碍。不同类型的深度学习架构,如卷积神经网络(CNN)、递归神经网络(RNN)、长/短期记忆(LSTM)、自动编码器(AE)和生成对抗网络(GAN),已应用于各种组学研究。
它在乳腺癌分类、自动青光眼检测、人类步态识别和智能融合辅助皮肤病变定位和分类方面优于许多传统的机器学习技术。所有这些分析都可以潜在地帮助医生提供精确的诊断和个性化的治疗。
随着深度学习的普及,Shaham 团队使用 ResNet 来去除批处理效应。虽然深度学习具有逼近高度非线性映射的强大能力,但 Shaham 团队的解决方案本质上是无监督的(即,不知道样本的疾病标签)。
另一方面,基于 GAN 的 NormAE 在非线性 AE 以消除批次效应和判别器之间构建对抗性训练过程以对批次标签进行分类。学习网络的辨别力对于代谢生物标志物的诊断和识别至关重要。不幸的是,仅通过减少不同批次之间的不匹配,诊断效果不一定会提高。
只有正确处理样本数据之间的批次效应,后续基于学习的分类和诊断才能受益。为了解决上述问题,上海交大的研究人员针对基于 MALDI MS 的代谢组学引入了一个联合深度学习框架,首先校准批次效应,然后进行样本分类(例如,得出疾病诊断)。
该框架由三个相互密切交互的主要网络组成:(1)给定单个输入批次的代谢组学数据,将它们通过校准器,使它们在潜在特征空间中对齐;(2) 一个后续的判别器从潜在空间中推导出来,在训练中受某个batch的已知标签监督,完成对另一个测试batch的分类;(3) 重构器也源自潜在特征空间并恢复所有输入批次,以确保输入批次在整个网络中得到很好的学习。
实验证明,该方法可以有效地消除批次效应,并且在分类准确性方面优于所有比较方法。这归因于该框架中相互交互的多个模块。对此,多重损失通过校准器 C 和鉴别器 D 生效,其中前者惩罚不同批次的域不匹配,后者奖励不同批次中相同类别的相似性。这两个任务的联合学习可以大大提高网络的整体性能。
已经开发了相当多的计算方法来消除批次效应,特别是在基因组学和转录组学领域。然而,它们在提高基于 MALDI MS 的代谢组学的诊断准确性方面可能不太有效。以 ComBat 为例,在实验中,准确率基本保持在原始数据的水平。fSVA 的算法最初是针对预测或分类任务而设计的,因此它可以产生比 ComBat 或 ratio_G 更好的性能。
虽然 ResNet 是基于深度学习的,但在准确率上并不优于 fSVA,如表 5 所示。一个可能的原因是它没有充分利用源标签提供的监督。至于 NormAE,除了在对抗过程中区分批次标签而不是生物标签外,潜在空间的维度也可能是它不如新方法的原因之一。
与其策略不同,隐藏层中从输入到输出的节点数量保持不变,尽管研究人员也使用了自动编码器骨干网。经过试验,压缩潜在空间中的特征数量会导致分类性能下降。总体而言,以前的方法不能有效地解决批次效应,尤其是在分类需要方面。
关于这项工作的实验结果有很多发现。鉴于 CyTOF 数据模拟研究的分类结果,人们可能会注意到患者 1 的批次内十倍交叉验证产生的 AUC 为 0.961。虽然在批内验证中完全排除了批效应,但应将这四个指标视为跨批验证的性能上限。然而,当将第 2 天作为源并将第 1 天作为目标时,跨批次 AUC 为 0.968,甚至超出了批次内上限。这种现象也可能是两批样本量不平衡造成的。换句话说,尽管存在批次效应,更大的训练数据量将有助于构建更健壮的分类器。
尽管该方法在所有比较指标中都取得了最佳性能,但这项工作仍然存在一些不可低估的不足。首先是研究人员可能无法通过枚举所有超参数来为每个比较算法获得完美的性能。
另外,他们的工作仅适用于单源单目标场景。在现实世界中经常遇到的多个批次的情况下,人们非常期望将训练好的模型调整为供多个未标记的目标批次使用,或者通过使用多个源批次进行训练来提高分类性能。
最后但同样重要的是,除了这种方法不再是无监督的并且需要领域知识之外,实现合理性能可能需要的标记数据量可能很大。未来的改进将从这些项目的角度进行。
这种新工具将产生巨大的潜力,其中其他代谢组学(即 LC-MS)甚至其他组学技术在临床研究中实际应用于大样本分析。
![]()